Scientific Achievement
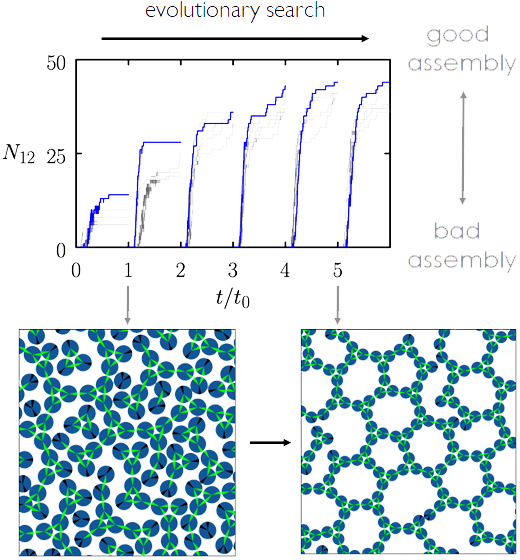
Neural networks were designed to autonomously control experimental parameters, such as temperature and chemical potential, in order to promote the self-assembly of target structures, finding new and nonintuitive pathways to high-quality molecular assemblies.
Significance and Impact
This study is the first to show that self-assembly protocols can be designed autonomously without continuously monitoring the structure during synthesis. This machine learning approach can inform any assembly protocol, including those not well-described by existing theories, such as the crystallization of pharmaceuticals.
Research Details
- The team designed a neural network scheme to control experimental self-assembly parameters using stochastic molecular simulations of prototypical “patchy” particles. Beyond the specification of the target, no human input was required.
- The team compared results from their evolutionary schemes to intuitive cooling simulations, and found that their networks were able to promote the self-assembly of desired structures faster and with higher fidelity.
- In the case where two possible structures have the same energy and are thus equally likely to occur in conventional simulations, the team’s neural networks could reliably promote one polymorph and suppress the other, offering new physical insights into these nonequilibrium processes.

